

Western blotting is a staple technique of the molecular biology lab. The robust nature of the antigen-antibody interaction allows the presence of specific proteins and peptides to be detected from complex mixtures. If the Western blot is not behaving as expected, our troubleshooting guide may help isolate the problem.
No bands are visible on the blotting membrane |
|
|
Can the protein marker be seen on the membrane? |
The prestained protein marker or ladder should be visible on the membrane after transfer. If not, then the transfer of the proteins from the electrophoresis gel to blotting membrane may have been unsuccessful. To confirm the transfer of proteins from the gel onto the blotting membrane, Ponceau S reversible stain can be a used before the blocking step. Although Ponceau cannot be used to identify a specific protein of interest, the presence of many faint pink/red bands on the blotting membrane confirms that proteins have been separated through the gel and have transferred onto the membrane. Small proteins may pass through the membrane or large proteins may fail to transfer from gel to membrane. Optimization of blotting conditions may be required, reducing or increasing the duration of transfer depending on the size of proteins to be blotted. If it is suspected that large proteins have not successfully transferred from gel to membrane, Coomassie stain may be used to detect proteins remaining in the acrylamide gel. Small proteins can sometimes be captured by the addition of a second membrane when blotting. |
|
Are air bubbles trapped between the gel and the membrane when blotting? |
The Ponceau stain will identify bubbles which have interfered with protein transfer, and appear as blank circles among the transferred proteins. Make sure to expel air from between the gel and membrane before transfer. |
Protein transfer confirmed. No specific protein bands visible. |
|
|
Does the primary antibody recognize the protein of interest? |
Check with the manufacturer of the primary antibody that it has been validated for detection of protein or epitope tag of interest by Western blot. If possible run a positive control for the protein of interest to confirm the specificity of primary antibody |
|
Are the primary and secondary antibody compatible? |
Use a secondary antibody which is specific for the species the primary was raised in, i.e. for detecting a rabbit primary use anti-rabbit secondary. |
|
Is the concentration of the antibody too low? |
Increase concentration of primary and/or secondary antibody, or incubate overnight at +4oC. |
|
Is the secondary antibody diluent appropriate? |
PBS/Tween 20 (0.05%) or TBS/Tween, without carrier proteins, is recommended as the secondary antibody diluent. Especially when using anti-goat or -sheep secondaries, avoid using milk or BSA in the diluent buffer. Bovine IgG may interact with the antibody due to homologous epitopes of the related species. |
|
Is there sufficient antigen present in the sample for detection? |
The detection limit of a Western blot is determined by a number of factors, but it is important to make sure that there is enough protein for detection in the sample prior to loading. Use a Bradford assay or a spectrophotometer at 280 nm to confirm total sample protein concentration, although this does not confirm the concentration of an analyte. If possible use a positive control to validate the experiment. If the protein of interest is very sparse, it may be necessary to enrich it in the sample using a technique such as immunoprecipitation (IP). |
|
Has the epitope tag been cleaved and lost from the protein during purification or sample preparation? |
Proteins can be subject to degradation and proteolysis during purification which may liberate an epitope tag from the protein of interest – making it impossible to detect through the epitope tag. The liberated tag may be detected as a smaller band than expected for the fusion protein. |
|
Has the primary antibody has been subjected to too many freeze/thaw cycles? |
Or has the primary antibody been re-used, reducing concentration? We recommend preparing fresh working dilutions on the day of use for both primary and secondary antibody. |
|
Has the reporter enzyme been inactivated? |
Be sure wash buffers and secondary antibody diluent are free of sodium azide, which strongly inhibits peroxidase activity. Glycerol lower in purity than ACS grade (>99.5%) also inhibits peroxidase activity. |
|
To confirm reporter enzyme activity, add a small sample of conjugated secondary directly to substrate and observe expected reaction. |
|
|
Is the ECL reagent out of date? |
If in doubt use fresh substrate.
|
|
Are the photographic developing reagents active? |
When using x-ray film to capture ECL signal it is important to check that reagents used to develop the film are still active. To confirm, expose the film to light and develop. |
Too much signal – signal from protein of interest obscured by high background. |
|
|
Has the antibody conjugate been titrated properly? |
Try diluting the conjugate further to reduce background. |
|
Has the gel been overloaded with protein? |
Loading amounts in excess of 10 μg per lane may create conditions leading to background staining. Measure protein concentration prior to loading. |
|
Is immunoprecipitation (IP) required to reduce total protein load? |
If the protein of interest is poorly expressed in the sample, IP can enrich its presence and eliminate extraneous proteins that contribute to background staining. |
|
Has blocking of the membrane been effective? |
Normal serum from the host species of the labeled antibody (5% v/v) is an excellent block, although 5% non-fat milk and 3% BSA are commonly used and may also be effective. Avoid milk or BSA when using primary antibody derived from goat or sheep. |
|
Does the secondary antibody bind the blocking reagent? |
Avoid using milk or BSA in the diluent when using a primary antibody derived in goat or sheep. Addition of a detergent such as Tween 20 may reduce weak binding. However using an alternative blocking reagent may be more appropriate for the antibodies used. |
|
Dilute antibodies in PBS/Tween, as proteins in diluent can aggregate and form sticky complexes which creates background. |
|
|
Was the washing step sufficient to remove unbound antibodies? |
Ensure the washing protocol includes sufficient volume, wash time and number of buffer changes. The wash buffer should include a detergent such as Tween 20 (0.05%). |
|
Is the blotting membrane contributing to background? |
Handle the blotting membrane as little as possible and use gloves and/or tweezers. Avoid letting membrane dry out during incubation steps. Nitrocellulose may have less auto–fluorescence than PVDF when conducting a fluorescent Western blot. |
|
Is the ECL blot in negative – black membrane with white bands? |
Antibody or antigen concentration is too high. Titrate antibody and sample to optimize. |
|
Observe unstained membrane in Western blot detection instrument to confirm autofluorescence. |
|
Unexpected bands detected – multiple signals can make interpretation of a Western blot difficult. |
|
|
Are bands detected at multiple molecular weights? |
Endogenous proteins can bind some primary antibodies non-specifically. For example, anti-His6 will detect some heat shock proteins (HSP). |
|
Are bands running at unexpected heights? |
Truncations, degradation and post-translational modifications can all change the expected migration of a protein. Make sure sample handling is optimal. Expression time courses, buffer composition and additives such as protease inhibitors or glycosylases can all help isolate where problems originate. If possible use a positive control. |
|
If possible run a negative control such as a non-transfected cell lysate/supernatant to confirm what the primary antibody is binding to the protein of interest not endogenous proteins in the sample. |
|
|
Protein degradation can lead to multiple bands if degradation is proximal to the epitope tag! |
The truncated proteins may be detected due to remaining epitope tag or remaining sequence sufficient for recognition by primary antibody. Protease inhibitors added to the sample can abrogate the effects of endogenous proteases in the sample. |
|
Is there heterogeneity in the protein of interest? |
Heterogeneity in post translational modifications to the protein of interest can result in multiple bands due to variations in migration for the same protein. Glycosylation, ubiquitination and phosphorylation can all add mass which can be detected by SDS-PAGE analysis (Mann and Jensen). |
|
Has the sample been completely reduced? |
Incomplete reduction of sample can lead to bands containing high order species which appear as bands at unexpected sizes or as smears. Use fresh reducing agent such as BME or DTT in sample loading buffer and boil in SDS (5-10 minutes). |
|
Has the sample been contaminated? |
Although unlikely, keratin and bacterial growth in running buffers can lead to unexpected bands on gel. Always handle gels wearing gloves and dispose of buffers containing growths. |
|
Run secondary antibody control without primary to confirm specific binding of primary antibody. |
|
|
It’s good practice to titrate antibody concentrations, thereby optimizing the signal to noise ratio. |
|
Uneven bands – Protein bands appear uneven. |
|
|
Are bands “smiling”? |
This can happen when lanes are overloaded or the buffer concentration isn’t ideal which causes the Western blot to run “hot”. Reducing the voltage, loading smaller volumes/ concentrations, adjusting buffer concentrations and running on ice can improve appearance. |
|
Do bands run unevenly in lanes? |
Improper casting of gels can lead to poor protein migration. Alteration of the gel casting protocol may be needed to encourage complete polymerization of acrylamide. Buying pre-cast gels can improve reproducibility. |
|
Is the blot unevenly stained? |
Use a shaker or a roller to encourage even distribution of all reagents. Insufficient incubation times may also contribute to uneven staining of the membrane. |
Western blotting after immunoprecipitation (IP). |
|
|
Are bands obscuring proteins of interest in the 50 or 25 kDa range? |
When performing a Western blot after IP, an anti-IgG (H+L) secondary antibody detects two bands corresponding to the heavy and light chains of the IP antibody (Figure 1A). Instead, use an anti-IgG, Light Chain Specific secondary antibody to avoid detection of the IP antibody heavy chain at 50 kDa (Figure 1B). |
|
Is the IP antibody from a different host animal than the primary (probing) antibody? |
If possible, use an antibody from one species for IP and then probe using a primary antibody from another species. Detect with a secondary antibody that is specific for the primary and cross-adsorbed against the IP species. |
|
Is the IP antibody from the same host animal as the primary (probing) antibody? |
Secondary antibody detection of the heavy chain of the IP antibody may obscure detection of a protein of interest in the 50 kDa range. Use an anti-IgG, Light Chain Specific secondary antibody to avoid recognizing the heavy chain of the primary antibody (Figure 1 (B)). |
|
Does the anti-IgG, Light Chain Specific antibody appear to detect the heavy chain? |
An overwhelming amount of IP antibody can trap the detecting antibody non-specifically. IP with minimal amount of antibody to prevent overloading on gel, and use an extended incubation if necessary. |
|
We recommend using a maximum of 10-20ug of IP antibody per lane/well to avoid overloading. |
|
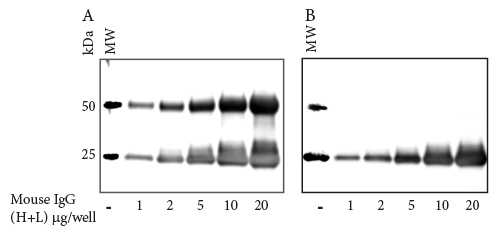
Figure 1: Western blotting after IP – Anti-Light Chain specific antibodies to avoid obscuring analytes in the 50 kDa range.
Gels were loaded with Mouse IgG, whole molecule (015-000-003)
After SDS-PAGE and transfer to nitrocellulose, blots were blocked with BSA (10% w/v). After incubation with secondary antibody, blots were developed with ECL substrate. Blots were imaged simultaneously, with auto exposure time based on bright bands.
A: The gel was probed using Goat Anti-Mouse IgG (H+L) (115-035-003), revealing bands corresponding to both heavy chains (50 kDa) and light chains (25 kDa).
B: The gel was probed with Goat Anti-Mouse IgG, Light Chain Specific (115-035-174), revealing only the 25 kDa band corresponding to Ig light chains.The IP antibody heavy chain is not detected, allowing visualization of the protein of interest near 50 kDa.
Light Chain Specific antibodies are available directed against goat, mouse, rabbit, rat and sheep see here.
If you are still experiencing problems with your Western blotting, our technical team would be delighted to help you problem solve. You can email them here.
References:
Alberts B et al (1994) Molecular biology of the Cell. 3rd Ed. Garland press. London
Kalyuzhny A (2016) Immunohistochemistry – Essential Elements and Beyond. Springer International Publishing Switzerland.
Technical Resources | About us | Contact us | Bulk Service
Licenses | Conditions of Use | Privacy Policy | Ordering Information

FM 545248