
In 1979, Towbin et al. first detailed the process of immunoblotting – the separation of proteins from a complex mixture using electrophoresis, followed by immobilization on a matrix and detection of the target protein through specific antibody-antigen binding. In 1981, Burnette et al. published an updated version and called the technique “western blotting.” In Part 1 of our Western blotting guide we will cover the following topics.

Overview
Introduction
Western blotting is widely used today in molecular biology, biochemistry and cell biology to detect specific proteins of interest from biological samples. This technique provides information about the target protein(s), including abundance and molecular weight, and provides a way of detecting protein-protein interactions and post-translational modifications.
Western blotting is performed in six stages:
- Sample preparation
- Protein separation by gel electrophoresis
- Protein transfer (electroblotting) onto a membrane
- Membrane blocking
- Immunodetection
- Visualization
Western blotting can be used to detect proteins from both natural and synthetic sources. Recombinant proteins from expression systems and endogenous proteins from biological samples can all be detected with western blot.
For proteins to be analyzed by western blot, the sample containing them must first be processed to make the target proteins accessible for analysis. For example, when screening for expression, proteins are extracted by lysing cells to release the proteins, so they can enter the separation matrix.
Protein extraction may be followed by denaturation and reduction to convert the proteins to their primary structure, which allows them to be separated by their molecular weight during electrophoresis.
Sodium dodecyl sulfate-polyacrylamide denaturing gel electrophoresis (SDS-PAGE) uses SDS to coat the proteins, masking their intrinsic charge and imparting an overall uniform charge proportional to their size.
The sample is then loaded into the gel, and a voltage is applied. The proteins migrate towards the positive electrode. The density of the gel retards their migration, allowing them to be separated by size. Following separation, the proteins are transferred (electroblotted) onto a membrane by placing the gel and membrane in direct contact and applying an electric field to pull the proteins into the matrix.
Protein transfer is followed by a blocking step, which uses a protein solution to minimize non-specific interactions. The membrane is then incubated with a primary antibody that specifically binds the protein of interest.
Following a further blocking step, a conjugated secondary antibody binds the primary antibody allowing the target protein to be visualized.
Either colorimetric or chemiluminescent substrates can be used to visualize signal from reporter enzyme conjugates. Signal from fluorescent dye conjugates is detected using an imaging device and can be used to detect multiple targets simultaneously.
Western blotting is a routine laboratory procedure that enables the detection and analysis of many proteins and their interactions. The success of western blotting hinges on the specificity of the antibody-antigen interaction and the potential for signal amplification with the addition of a secondary antibody. These factors make western blotting extremely sensitive, enabling the detection of proteins down to picogram levels. Depending on the type of reagents and detection method, semi-quantitative and quantitative results can be obtained.
Western blotting can be performed efficiently and reliably to produce high-quality blots. This guide will assist you with selecting the correct reagents, picking suitable antibodies and detection methods, and troubleshooting your experiments.
Sample Preparation
Accurate separation of proteins during electrophoresis is a crucial step in the western blot. It relies on the sample being processed correctly so that the protein(s) of interest are in a form where they can migrate through the gel.
Proteins from a variety of sources can be analyzed by western blot. Samples may be derived from protein expression systems such as mammalian or bacterial cell culture, or from clinical tissue samples, each requiring different processing to produce quality protein for their ultimate use. These protein extraction methods are detailed in Table (1); however, for western blotting it is often not necessary to process samples so strictly.
Due to the specificity of immunoblotting, samples for western blotting can be crudely processed so that proteins can be extracted and transferred into a solution suitable to introduce the sample into the separation gel for electrophoresis. Crude samples, in particular, may be viscous due to the presence of polymeric biomolecules, such as DNA, and gel loading may be negatively impacted.
Example sample preparation process for bacterial expression system.
- Take a 1 ml sample of E.coli cell culture and transfer to a microcentrifuge tube on ice.
- Spin for 20 mins at 13,000 rpm at 4°C.
- Discard the supernatant.
- Resuspend cells in 50 µl loading buffer and boil for 5 mins at 100°C.
- Centrifuge at 13,000 rpm for 5 mins.
- Load sample.
Example sample preparation of adherent cells, e.g., mammalian expression system (HEK293).
- Place the tissue culture plate on ice and wash the cells with ice-cold PBS.
- Aspirate the PBS, then add an appropriate volume of lysis buffer (e.g. 1 mL per 107 cells/100 mm dish 150 cm² flask; 0.5 mL per 5×106 cells/60 mm dish/75 cm² flask).
- Detach cells from the well/flask using a cell scraper, then agitate with a pipette tip or through trypsinization.
- The cell suspension is transferred into a pre-cooled microcentrifuge tube.
- The sample is then microcentrifuged at 4°C. Conditions will depend on the cell type and should be determined for your experimental setup. E.g., HEK 293 may tolerate 15 mins at 1000 rpm.
- The supernatant is aspirated from the sedimented cells and transferred to a fresh tube on ice.
- Loading dye is added to the supernatant and boiled for 5 mins at 100°C.
- The cell pellet may also have loaded dye run alongside the supernatant.
The sample is spun for 5 mins at 13,000 rpm before loading into the gel for electrophoresis.
Lysis and Solubilization
To enable proteins to enter the separation gel, they must be in a soluble form. The first stage of sample preparation is lysis.
Intracellularly expressed proteins are extracted by lysis, which ruptures the cell’s plasma membrane and/or cell wall, to release the protein. Proteins expressed extracellularly (secreted) do not require lysis, although cells may be processed and loaded to observe any protein trapped due to improper synthesis.
Large Scale Protein Purification – Extraction Methods
Proteins from a variety of sources can be analyzed by western blot. Samples may be derived from protein expression systems such as mammalian or bacterial cell culture, or from clinical tissue samples, each requiring different processing to produce useful blots.
The method used for protein extraction depends on the nature of the sample. Protein expression is performed in different cell types such as mammalian, insect, bacterial, and yeast, and some samples may come from tissues; these have structural properties that require different processing.
Secreted proteinsSome proteins expressed in mammalian or insect cell systems may also express in the supernatant and do not need to be retrieved from cells. A good tip is to keep a sample of the supernatant and load that onto the gel alongside the cell lysate when screening for expression. Screening and ProcessingProtein expression is commonly screened by western blotting. For example, the expression of several clones may be compared between each other, or their expression in different expression systems. In these screening trials, a sample from the protein prep is taken and loaded for analysis. Often this will compare the supernatant to the cell pellet to localize where expression occurs. Total protein seen by coomassie staining may be compared to the expression of the protein of interest picked up specifically in the western blot. The rudimentary processing undertaken in the screening trial may not be representative of how the final expression (prep) is processed for its ultimate use, such as the careful purification steps needed for crystallographic trials. 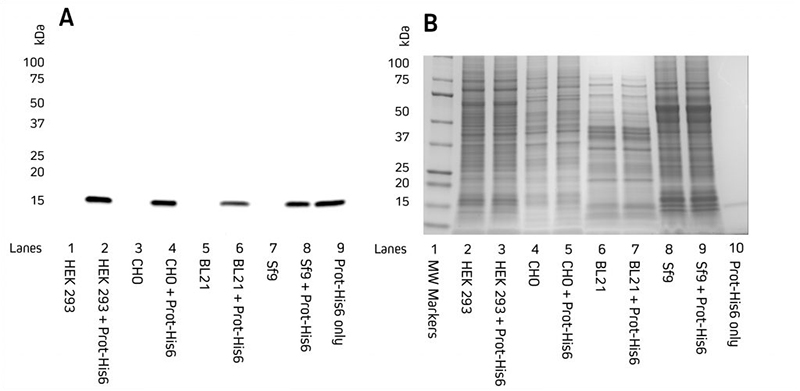 A. Western blot. Gels were electroblotted onto nitrocellulose membrane, blocked with 5% BSA in PBS/0.2% Tween 20, and probed with Jackson ImmunoResearch’s HRP Rabbit Anti-His Tag (300-035-240) at 1:20K dilution and visualized using a digital imager. B. Gels were stained with Coomassie. |
| Lysis Method | Mode of Action | Suitable For |
|---|---|---|
| Freeze-thaw | Repeated rapid freezing using liquid nitrogen and thawing. The formation of ice crystals disrupts the cell membranes and releases the proteins. | Mammalian and insect cell cultures |
| Osmotic | Repeated transfer between high- and low-osmotic environments to disrupt the cell membranes and release proteins. | Mammalian and bacterial periplasmic fractions |
| Ultrasonication | Disrupts the cell membranes using high-frequency sound waves. Relatively simple and low cost. However, ultrasonication also produces heat, which must be controlled by cooling the sample on ice in between sonication. | E.coli, Mammalian
and Insect cell cultures |
| Enzymatic digestion | Lysozyme to break down the rigid, carbohydrate-rich cell wall. | Bacterial, yeast and plant cultures |
| Homogenization (mechanical) | Mechanical mincing of the sample in a chilled buffer, typically with a Waring blender or Polytron, disrupts the cell membranes with liquid shear forces. May also include glass beads of various sizes to promote shearing. Mechanical grinding (e.g., mortar and pestle), sometimes at ultra-low temperatures (liquid nitrogen). | Mammalian and plant tissues
Bacterial and yeast cell cultures |
| Decompression | Equilibrating the cells at high pressure (~5500 kPa) before transferring the sample to a low-pressure chamber. The rapid pressure drop bursts the cells. High pressure can be induced mechanically in a press or with an inert gas, such as nitrogen. | Bacterial cultures |
| Detergent | Centrifugation of the cell sample followed by resuspension in a lysis buffer containing a detergent. This detergent compromises the cell membranes’ integrity, causing the lysis of the cells and releasing the proteins. | Mammalian, insect and bacterial cultures |
Lysis Buffers
The purpose of the lysis buffer is to rupture the cell membrane to release the protein. Lysis buffers commonly include detergents that disrupt the lipid bilayer of cell membranes and form micelles. There are a number of buffer formulations that have been validated for use with different cell types or protein expression locations, such as RIPA or NP-40, which contain detergent. The processing of materials should be performed on ice to minimize degradation and denaturation from cellular components that may alter the protein.
Proteases and their Inhibition
Solubilization methods, such as processes involving mechanical disruption to the cells, result in the release of intracellular proteases. These proteases can digest and truncate the protein of interest, which may cause multiple bands to be observed on the blot.
The susceptibility of proteins to proteases varies depending on their amino acid composition, with intracellular proteins being less resistant than cell-surface or extracellular proteins. Membrane proteins, when solubilized by detergents, are particularly susceptible to protease degradation. Bioinformatic tools are available to predict the susceptibility of a protein to proteolysis in advance. Protease inhibitors may be used to prevent proteolysis; these can be cocktails of chemical and enzymatic inhibitors that may be added to the lysis buffer before addition to the cells or to the buffer the protein is to be stored in.
There are a range of inhibitors that prevent the common classes of serine-, cysteine-, aspartic-proteases, as well as aminopeptidases and metalloproteases. Different expression systems or susceptibility to protein activity inhibition may prevent the use of certain inhibitors. Proprietary mixtures of protease inhibitors are often available, or single agents can be applied.
Azide may be used with care in buffers to prevent bacterial growth, which can also be a source of proteases. Dephosphorylation may occur after the cells have been lysed. If phosphorylated proteins are the target of western blotting, phosphatase inhibitors, such as sodium vanadate, should also be added to the lysis buffer. Samples should be kept on ice throughout the sample preparation to minimize the possibility of degradation and dephosphorylation.
| Inhibitor | Protease | Cautions |
|---|---|---|
| EDTA (Ethylenediaminetetraacetic acid) | Metalloproteases | Incompatible with some affinity chromatography columns and may inhibit enyzmes requiring divalent cations. |
| Pepstatin | Acid proteases – pepsin | |
| Leupeptin | Serine and Cysteine proteases | Toxic. |
| TLCK (nα-tosyl-l-lysine chloromethyl ketone hydrochloride) | Serine proteases (Trypsin-like proteases) | Noxious/Foul Odor |
| PMSF (Phenylmethylsulfonyl fluoride), AEBSF is a common water soluble version and is more stable. | Serine proteases | Neurotoxin. Short half life—prepare fresh. |
| Azide | Bacterial growth | Can interfere with downstream processing/detection. Poisonous. |
| Sodium orthovanadate | ATPase, alkaline phosphatase and tyrosine phosphatases | Toxic. |
| Sodium pyrophosphate | Serine/threonine phosphatases. | Corrosive and irritant. |
pH
The pH of the lysis buffer is important to control during sample preparation to ensure the stability and activity of the protein are maintained. The pH dependency of the activity and stability of a protein can be plotted to generate a typically bell-shaped curve of which the maximum (maxima) is termed the pH-optimum; this can be used as a metric for the pH-dependent properties of the protein. By using a buffer at the correct pH, protein stability and solubility are ensured, preventing aggregation and precipitation. Allied with stability is the protein’s activity. Using the correct pH ensures the functionality of the protein is retained for downstream use.
By using a buffer at the correct pH, protein stability and solubility are ensured, preventing aggregation and precipitation. Allied with stability is the protein’s activity. Using the correct pH ensures the functionality of the protein is retained for downstream use.
The pH of the buffer is typically one pH unit above or below the isoelectric point of the protein, commonly in the physiological range between pH 6 and 8. pH is maintained using buffering systems containing the combination of a weak acid and its conjugate base, or a weak base and its conjugate acid. Many systems are available to generate buffer solutions for a range of pHs and some are listed in figure 1. It is important to note that pH of a buffer can be temperature-dependent which is why electrophoresis and blotting apparatus incorporate a cooling component.
| Buffer | pH range |
|---|---|
| Citric acid – NaOH | 2.2 – 6.5 |
| Sodium citrate – citric acid | 3.0 – 6.2 |
| Sodium acetate – acetic acid | 3.6 – 5.6 |
| Cacodylic acid sodium salt – HCl | 5.0 – 7.4 |
| MES – NaOH
(2-(N-morpholino)ethanesulfonic acid) |
5.6 – 6.8 |
| Sodium dihydrogen phosphate – disodium hydrogen phosphate | 5.8 – 8.0 |
| Imidazole – HCl | 6.2 – 7.8 |
| MOPS – KOH (3-(N-morpholino)propanesulfonic acid) | 6.6 – 7.8 |
| Triethanolamine hydrochloride – NaOH | 6.8 – 8.8 |
| Tris – HCl ( Tris(hydroxymethyl)aminomethane) | 7.0 – 9.0 |
| HEPES – NaOH
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid ) |
7.2 – 8.2 |
| Tricine – NaOH | 7.6 – 8.6 |
| Sodium tetraborate – boric acid | 7.6 – 9.2 |
| Bicine – NaOH | 7.7 – 8.9 |
| Glycine – NaOH | 8.6 – 10.6 |
Detergents
Non-ionic detergents can be used to increase the solubility of non-polar, insoluble proteins. Examples of these are Triton™ X-100 and Tween(r) – 20.
Osmotic stabilizers
When purifying proteins from specific subcellular structures, such as organelles from Eukaryotic cells, lipid membranes, or proteins from the periplasmic space of bacteria, osmotic stabilizers such as high concentrations of sucrose are often included during the cell lysis step. Their inclusion helps to stabilize sub-cellular structure during lysis and can also prevent cell lysis during cell wall degradation.
Subsequent centrifugation at specific g-forces unique to the subcellular structure being isolated and the inclusion of density gradients (e.g., sucrose or glycerol fractionation) are often used to further enrich proteins from subcellular structures before western blotting.
Presence of osmotic stabilizers may affect how a protein migrates on a gel, and samples may need to be diluted into different osmotic strength buffers before loading.
Salts
Salts are added to modify the ionic strength of the buffer solution. Adding salt can improve protein solubility; however, too much salt can decrease solubility and precipitate the protein.
By optimizing the ionic strength of a solution, proteins can be encouraged to retain their folded conformations, which in turn can deter damage from proteolysis by preventing exposure of vulnerable internal residues. Stabilization of surface charge discourages protein aggregation.
Sample clarification
Centrifugation is used to clarify samples. It may be used following lysis, whereby the cell lysate is ultracentrifuged at very high speeds for extended periods of time to pellet the cell debris and chromosomal DNA, which is then discarded and the solution containing the solubilized proteins are retained.
For proteins expressed extracellularly into culture medium, for example, in mammalian cells, the speed and duration of centrifugation must be modified to provide less damaging conditions to prevent lysis of the cells. Centrifugation, often through density gradients, is also used to separate cell fractions.
For example, microsomes (endoplasmic reticulum, plasma membrane) can be pelleted at high speeds after initial clarification by centrifugation at lower speeds.
Remnants of cellular components from improperly clarified lysates and high NaCl concentrations can result in improper separation of the proteins within a sample during SDS-PAGE, causing blurred bands or proteins resolving at unexpected molecular weights.
The presence of nucleic acids can also clog the wells and affect sample migration, so deoxyribonuclease (DNase) can be added to eliminate this contaminant. Dialysis or gel filtration can also be performed to reduce the NaCl concentration. These cleanup techniques enable a more accurate separation during electrophoresis.
Sample Concentration
The total protein concentration of the sample ideally should be determined before loading, using methods such as the Bradford, Lowry or bicinchoninic acid (BCA) assays. Predetermining sample concentrations enables wells in the separation gel to be loaded with equal volumes and concentrations to optimize consistency across the gel. If too much protein is loaded into a well, it can influence the migration of proteins in neighboring lanes, making it difficult to interpret results.
Depending on the size of the wells, 10-50 µgs total protein per lane should be adequate. However, this may be too concentrated if using pure protein.
Low abundance proteins may need to be concentrated to enable detection by western blot analysis. Purification tags such as HIS6 tags allow proteins to be concentrated using affinity columns such as nickel. It is important to make sure that the elution buffer is compatible with the loading buffer. Alternatively, where antibodies specific to the protein of interest are available, methods such as immunoprecipitation can be used to effectively concentrate proteins.
Protein ConcentrationLarge protein preps commonly require a concentration step during purification steps to reduce material volumes to a more manageable size. Affinity, ion exchange and metal chromatography, or tangential flow filtration can be used to remove excess liquid volumes and concentrate either total or target proteins. Western blotting samples may not require a concentration step, as abundance of the protein is unlikely to be below the limit of detection due to the sensitivity of the technique. |
Optimal protein resolutionLoading too much protein in a well can cause proteins to migrate unexpectedly, but so can an improper set up, such as when loading buffers have an excessively high concentration of salts or denaturants (e.g., guanidine HCl). In addition, gel running conditions that excessively increase temperature during the gel or western blot run (e.g., excessive voltage or salt concentration in running buffer) can negatively affect protein separation. See Chapter 4 for advice on how to troubleshoot your blot. |
| Effect | Cause | Solution |
|---|---|---|
| Gummy or viscous sample/ poor gel entry or migration | DNA | DNAse |
| Loading dye changing color | pH | Buffer exchange or addition of acid/base |
| Diffuse band | Low or high salt | Buffer exchange |
Loading Conditions/Buffers
Prior to loading, the sample is mixed with a loading buffer, which facilitates protein denaturation and the loading of the sample into the separation gel. It is usually a combination of dye, reducing and denaturing reagents and glycerol. The sample is then boiled for 5 mins at 100°C and then centrifuged briefly before loading. In some situations, for instance, with highly hydrophobic integral membrane proteins, temperatures lower than 100°C are required.
Loading dyes
Loading dyes are added to the sample to visualize loading and sample migration during electrophoresis. A frequently used loading buffer, Laemmli loading buffer, is a combination of bromophenol blue, glycerol, Tris, SDS and a reducing agent such as DTT or BME. Because of its small size, bromophenol blue migrates faster than the samples’ proteins and provides a migration front to monitor the electrophoresis process and prevent sample run-off. It also goes yellow if acidic conditions are encountered, indicating a buffering system is not sufficient, or a processing step has been omitted. The glycerol makes the sample denser than the running buffer, enabling the sample to “sink” to the bottom of the well. After boiling the sample in the loading buffer, the sample is briefly centrifuged.
| Tris (pH 6.8 1M) | 10 ml |
| SDS | 4 g |
| Glycerol | 20 ml |
| BME | 10 ml |
| Bromophenol blue | 0.1 g |
| dH2O | 50 ml |
Reducing/Non-Reducing Conditions
Samples to be analyzed by western blot are denatured and reduced so that proteins resolve according to their molecular weight during electrophoresis. The addition of reducing agents, such as β-mercaptoethanol (BME) or dithiothreitol (DTT), reduces the disulfide bonds between cysteine residues. Treatment with SDS denatures the proteins by disrupting the non-covalent bonds within and between the residues, which linearizes the proteins into “floppy” chains of amino acids. SDS also effectively saturates the polyamide backbone of proteins, reducing the effect of charge on protein migration.
CautionIncomplete reduction of the sample may result in proteins not resolving as expected, which may be observed as smeared bands or bands of unexpected sizes. Therefore, fresh reducing agents should be added to the loading buffer on the day of use. Insufficient heating can also lead to incomplete denaturation of the sample. |
Native or Non-Reducing GelsNon-reducing or native gels are used to observe the native behavior of the proteins, such as their stoichiometry or interaction with other proteins in the solution. These proteins are usually visualized by Coomassie or silver staining rather than immunoblotting. In this instance, reducing agents should not be added to the loading buffer, SDS is removed from the loading and running buffers, and samples are not boiled. Table 5 details the components in the buffers depending on the protein state required. |
| Protein State | Sample Loading Buffer | Gel Running Buffer |
|---|---|---|
| Reduced and denatured | SDS
βME or DTT Boil 5–10 minutes* *for integral membrane proteins, lower temperature (e.g. 70 degrees C) may be preferred |
SDS |
| Reduced and native | βME or DTT | No SDS |
| Oxidized and denatured | SDS
Boil 5–10 minutes |
SDS |
| Oxidized and native | No SDS |
Bromophenol blue is also generally added to the sample loading buffer to enable visualization of protein migration during gel electrophoresis. Because of its small size, bromophenol blue migrates faster than the samples’ proteins and provides a migration front to monitor the electrophoresis process and prevent sample run-off.
Found Part 1 of our Western blotting guide useful? View part 2 here
References
- Blancher C. et al. (2001). SDS-PAGE and Western Blotting Techniques. Methods in Molecular Medicine. doi: 10.1385/1-59259136-1:145.
- Lee C. (2007). Protein Extraction from Mammalian Tissues. Methods in Molecular Biology. doi: 10.1007/978-1-59745-257-1_29
- Jackson ImmunoResearch Laboratories Inc. (2017). Western Blotting Troubleshooting Guide! https://www.jacksonimmuno.com/ secondary-antibody-resource/technical-tips/western-blot-trouble-shooting/
- GE Healthcare. (2011). Western Blotting Principles and Methods. https://www.sigmaaldrich.com/content/dam/sigma-aldrich/ docs/Sigma-Aldrich/General_Information/1/ge-western-blotting.pdf
- Najafov A. et al. (2017). Western Blotting Guru. Elsevier Inc. https://www.sciencedirect.com/book/9780128135372/westernblotting-guru
- Bass J.J. et al. (2017). An Overview of Technical Considerations for Western Blotting Applications to Physiological Research. Scandinavian Journal of Medicine and Science in Sports. doi: 10.1111/sms.12702
- Talley, K., & Alexov, E. (2010). On the pH-optimum of activity and stability of proteins. Proteins, 78(12), 2699–2706. https://doi. org/10.1002/prot.22786

| Learn more: | Do more: |
|---|---|
| Colorimetric western blotting | Spectra Viewer |
| Chemiluminescence western blotting | Antibodies for signal enhancement |
| Fluorescent western blotting | |
Technical Resources | About us | Contact us | Bulk Service
Licenses | Conditions of Use | Privacy Policy | Ordering Information

FM 545248