
Key stages in the ELISA protocol
A typical ELISA can be broken down into three main steps, namely capture, blocking, and detection/quantification. It also involves numerous wash steps to ensure any unbound reagents are removed from the microplate wells between steps.
Capture
Whichever ELISA format is chosen, assay setup begins with immobilizing the analyte or antibody on the microplate. The microplate will invariably be flat-bottomed and made of polystyrene, however, its color will be dictated by the detection method. Clear plates are used for colorimetric detection, black plates for fluorescent detection, and either black or white plates with clear bottoms are used for chemiluminescence. Microplates should always be sourced from a trusted supplier to avoid plate-to-plate variability or the issue of sticky plates which may exhibit excessive non-specific binding that contributes to background signal.
Immobilization involves diluting the analyte or antibody in an appropriate coating buffer and incubating it in the microplate wells to allow binding to occur via passive adsorption. 0.2M carbonate/bicarbonate at pH 8.4-9.6, depending on the protein isoelectric point, is the most common coating buffer, although PBS or TBS are sometimes used.
The main role of the coating buffer is to stabilize the analyte or antibody and conserve the competency of the antigenic regions while not inhibiting hydrophobic interactions between the protein and the polystyrene plate. It is also essential that the coating buffer is protein-free. Any other proteins present in the coating buffer could bind to the microplate wells and become a source of non-specific background signal.
Where oriented binding of an antibody is required, plates coated with Protein A or G may be a preferred choice. However, these should be avoided for sandwich ELISA because detection antibodies and secondary antibodies can also bind Protein A or G. Biotin- or streptavidin-coated plates can also be used to capture analytes or antibodies where these have been labeled with streptavidin or biotin, respectively.
In addition to choosing a suitable microplate and coating buffer, factors that require optimization include the incubation time/temperature for immobilization and the concentration of the biomolecule that will be used for coating. Once optimal conditions have been identified, these should be recorded to help ensure experimental consistency.
Sample Preparation
Samples analyzed by ELISA vary extensively and can include cell lysates, cell culture supernatants, tissue homogenates, and bodily fluids such as serum, urine, cerebrospinal fluid, tears, and saliva. ELISA is also used extensively to study foodstuffs such as milk and environmental material such as river water. While the sample preparation method will vary according to the material under investigation, it is essential that samples are handled carefully so that their integrity is preserved.
General good practice guidelines for sample handling and preparation often recommend that samples be stored at -80oC, although this varies depending on the nature of the sample material. Irrespective of the storage temperature, freeze-thaw cycles should always be kept to a minimum. It is also important to keep samples on ice when they are in use and to aliquot them prior to storage for greater consistency between experiments run on separate days.
The sample matrix is a key consideration when running an ELISA since this can often affect the assay readout. Spike and recovery experiments are a quick and easy way to assess matrix effects and can be used to determine whether controls and standards should be diluted in a matrix that more closely matches that of the sample material under investigation. Fats and lipids can especially confound ELISA readouts, and it may be necessary to remove them by solid phase extraction (SPE) prior to analysis.
Another factor that must be considered is the biosafety level of the samples being analyzed. Infectious material should be handled in an appropriate biological safety cabinet to contain any aerosols or droplets and suitable personal protective equipment (PPE) should always be worn.
Analyte-specific antibody considerations
Validation
Antibodies form the core of any ELISA, highlighting the critical importance of selecting well-validated antibody reagents to support your research. Antibodies raised against the analyte of interest must bind their target specifically and with high affinity and should always be proven to work in the ELISA application – both by the antibody manufacturer and through in-house validation – before being used with precious sample material.
Host species
The host species is a key consideration when choosing a primary antibody, not least because it will dictate the selection of an appropriate secondary antibody reagent; this must recognize the primary antibody host species and be conjugated to a suitable detection moiety. Sourcing antibodies from larger hosts such as goats or sheep can provide cost benefits due to the greater antibody volumes that are produced, while antibodies from camelid hosts provide the additional advantage of being better able to access cryptic epitopes as a result of their small size.
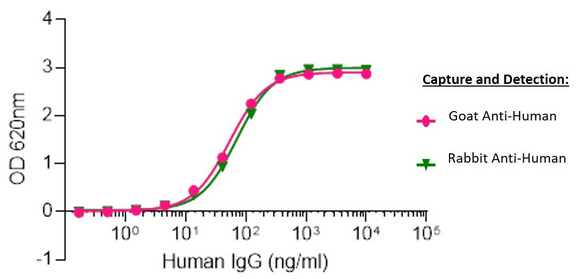
Sandwich Elisa: Elisa plates were coated with either AffiniPure Goat Anti-Human IgG, Fcγ fragment specific (109-005-008) or AffiniPure Rabbit Anti-Human IgG, Fcγ fragment specific (309-005-008).
After blocking with Bovine Serum Albumin (IgG-Free, Protease-Free) (001-000-162), ChromPure Human IgG, whole molecule (009-000-003) was then titrated across successive wells and the non-binding fraction was washed off. Captured IgG was detected with either Peroxidase AffiniPure Goat Anti-Human IgG, Fcγ fragment specific (109-035-008) or Peroxidase AffiniPure Rabbit Anti-Human IgG, Fcγ fragment specific (309-035-008) diluted 1:40,000 in assay buffer. Both Goat and Rabbit antibodies displayed almost identical limits of detection and signal strength in this assay.
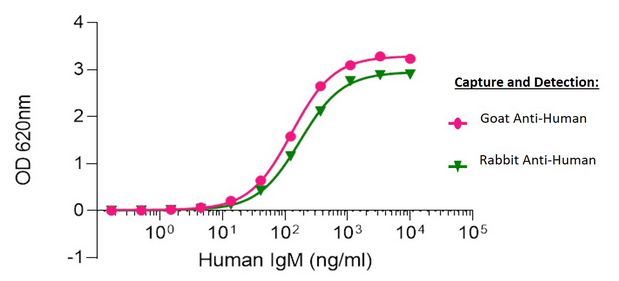
Sandwich Elisa: Elisa plates were coated with either AffiniPure Goat Anti-Human IgM, Fc5μ fragment specific (109-005-129) or AffiniPure Rabbit Anti-Human IgM, Fc5μ fragment specific (309-005-095). After blocking with Bovine Serum Albumin (IgG-Free, Protease-Free) (001-000-162), ChromPure Human IgM (myeloma), whole molecule (009-000-012) was then titrated across successive wells and the non-binding fraction was washed off. Captured IgM was detected with either Peroxidase AffiniPure Goat Anti-Human IgM, Fc5μ fragment specific (109-035-129) or Peroxidase AffiniPure Rabbit Anti-Human IgM, Fc5μ fragment specific (309-035-095) diluted 1:40,000 in assay buffer. Both Goat and Rabbit antibodies displayed similar limits of detection and have strong signals to noise.
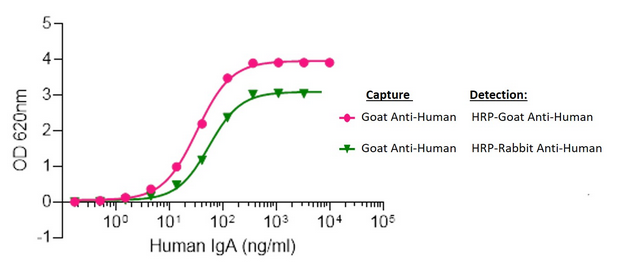
Elisa plates were coated with AffiniPure Goat Anti-Human Serum IgA, α chain specific (109-005-011). After blocking with Bovine Serum Albumin (IgG-Free, Protease-Free) (001-000-162), ChromPure Human Serum IgA, whole molecule (009-000-011) was then titrated across successive wells and the non-binding fraction was washed off. Captured IgA was detected with either Peroxidase AffiniPure Goat Anti-Human Serum IgA, α chain specific (109-035-011) or Peroxidase AffiniPure Rabbit Anti-Human Serum IgA, α chain specific (309-035-011) diluted 1:20,000 in assay buffer. Both Goat and Rabbit antibodies displayed similar limits of detection and high signal to noise.
Clonality
Researchers must also decide between using a monoclonal or a polyclonal antibody. While monoclonals offer more consistent lot-to-lot performance and guaranteed long-term supply, polyclonals can provide a level of signal amplification by recognizing multiple epitopes and may be less expensive. Polyclonals can also increase the likelihood of target detection in situations where the analyte is compromised or not as expected, and they typically become available sooner when a novel target is first discovered. When developing a sandwich ELISA, many researchers choose to use a monoclonal antibody for antigen capture and a polyclonal antibody for detection; where two monoclonals are used, it is vital that each recognizes a different epitope to prevent competition for target binding.
Format and Purity
Antibody format is a further consideration. Although many primary antibodies are purified by the manufacturer, others are supplied in the form of tissue culture supernatant or ascites fluid. As well as having a lower antibody concentration, unpurified antibody preparations can especially be challenging to work with where there is a requirement to conjugate the antibody in-house or coat onto an ELISA plate.
Secondary/Detection Antibody Considerations
Secondary or detection antibodies are equally as important as analyte-specific antibodies to producing meaningful ELISA results. Where they are employed for indirect detection purposes, it is essential that they are compatible with the host species of the analyte-specific antibodies.
Cross-Adsorption
Oftentimes, the experimental setup of the ELISA may necessitate the use of secondary antibodies that have been cross-adsorbed against other species present in the assay. Using cross-adsorbed antibodies minimizes the potential for cross-reactivity (e.g. secondary antibody binding to coating antibodies) that may cause background signal and impede assay performance and analysis.
Antibodies should always be handled with care and should be titrated during ELISA optimization to identify a dilution that achieves an acceptable signal window in the assay.
Blocking
Blocking is used to prevent non-specific binding that can cause background signal. Its importance should not be overlooked since non-specific binding can interfere with assay performance and subsequent data analysis. Blocking is performed after plate coating and before the sample is added to the microplate wells, and its efficiency can be established using appropriate controls, as discussed further on.
Bovine Serum Albumin (BSA) is widely used as a blocking agent for ELISA. However, it is important to be aware that many commercial BSA preparations contain contaminating bovine IgG that may become a target for antibodies with reactivity to bovine, goat or sheep material, resulting in background. Commercial BSA preparations may also contain proteases that can degrade both the sample material and the antibodies being used. These problems can be avoided by choosing a source of BSA that is certified as being IgG- and protease-free.
Normal serums (serums derived from non-immunized animals) are often used as alternative blocking reagents to BSA. They are strongly recommended to prevent antibody binding to conserved sequences and/or Fc-receptors as well as to reduce background resulting from non-specific binding. Best results are obtained using diluted (typically 5%v/v) normal serum derived from the same host species as the labeled antibody.
Caution: Blocking with normal serums can also be risky. If the primary binds to a serum protein with significant epitope identity to the host species of the serum then it will cause problems such as falsely elevated signal.
Conjugate
Another factor to consider during antibody selection – either when using directly-conjugated analyte-specific antibodies or secondary antibodies for detection – is the nature of the molecule used for visualization. This is typically an enzyme that provides a colorimetric or electrochemiluminescent readout, or a fluorescent dye, as discussed below. A suitable reader is critical for signal measurement.
Detection and Quantification
ELISA detection can be categorized as colorimetric, fluorometric or chemiluminescent. The chosen method is largely dictated by the level of sensitivity required, whether there is a need for multiplexing, and the reagents and instrumentation available.
During colorimetric detection, antibodies conjugated to enzymes such as horseradish peroxidase (HRP) or alkaline phosphatase (AP) are used to catalyze the conversion of a substrate into a soluble colored product. Some common colorimetric ELISA substrates and their stop solutions are shown in Table 1.
| Enzyme | Colorimetric substrate | Stop solution | Final color / read wavelength |
|---|---|---|---|
| Horseradish peroxidase (HRP) | 3,3′,5,5′-Tetramethylbenzidine (TMB) | 0.16M sulfuric acid | Yellow 450 nm |
| o-Phenylenediamine dihydrochloride (OPD) | 3M hydrochloric acid or 3M sulfuric acid | Orange 492 nm |
|
| 2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) | ABTS stop solution | Yellow 410 nm |
|
| Alkaline phosphatase (AP) | p-Nitrophenyl Phosphate (pNPP) | 1M sodium hydroxide | Yellow 410 nm |
The main advantages of colorimetric detection are that the readout is readily measured using a standard spectrophotometer, and the necessary enzyme-conjugated antibodies and their substrates are inexpensive and widely available. Disadvantages are that only a single target can be detected and the technique can be prone to operator bias; for example, the length of time allowed for color to develop can be highly subjective.
Fluorometric detection uses antibodies conjugated to fluorophores to produce a measurable fluorescent signal. Researchers occasionally refer to an ELISA using fluorometric detection as a FELISA or FluorISA. Popular fluorophores include fluorescent proteins such as R-phycoerythrin (R-PE), allophycocyanin (APC) or peridinin-chlorophyll-protein (PerCP), and fluorescent dyes such as fluorescein isothiocyanate (FITC) or Alexa Fluor®. Advantages of fluorometric detection are that it has a greater dynamic range than colorimetric detection and it also enables multiplexing. Disadvantages are the requirement for a reader fitted with the appropriate filters and the fact that some fluorescently-conjugated antibodies can be expensive. It is also necessary to take appropriate steps to prevent the fluorescent signal from being compromised; for example, by performing incubations in the dark.
Chemiluminescent detection typically relies on antibodies conjugated to HRP to catalyze a chemical reaction. This generates energy in the form of measurable light, commonly using a luminol-based substrate. Chemiluminescence is widely recognized for its exceptional sensitivity and its broad dynamic range. However, these benefits are counterbalanced by the need for specialized reagents and access to a dedicated reader.
Whether colorimetric, fluorometric or chemiluminescent detection is chosen, the amount of target analyte in each of the sample wells is quantified by correlating the ELISA readout to a standard curve generated on the same microplate. This is achieved by adding known quantities of the target analyte (the standard) to sequential wells, typically as a serial dilution, whereby the concentration of test samples is calculated by interpolation on the graph. Carefully selected controls are used to define the top and bottom of the standard curve and an analysis package is typically employed to ensure data is handled consistently.
Plate Washing
During an ELISA, plates are washed after coating, after sample incubation, and after every incubation step within the chosen detection method. It is common practice for at least 3 x 5-minute washes to be applied each time, with this being increased to 6 x 5-minute washes following the final incubation with the enzyme conjugate. Although Tris-buffered saline (TBS) and phosphate-buffered saline (PBS) containing 0.05% (v/v) Tween®-20 are usually used for washing during an ELISA, the wash buffer should always be optimized as part of assay development.
The ELISA Workflow
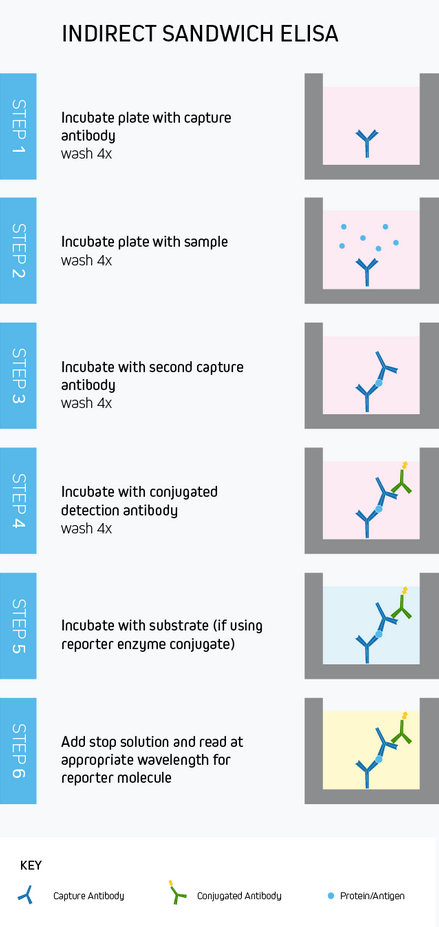
While capture, blocking, detection and quantification, and plate washing are common to every ELISA, the intricacies of the workflow will vary according to the assay format and whether direct or indirect detection is used. To provide an example, Figure 10 depicts a typical sandwich ELISA workflow with indirect colorimetric detection. Whichever method is chosen, rigorous optimization and validation are vital to generate meaningful results.

| Learn more: | Do more: |
|---|---|
| Colorimetric western blotting | Spectra Viewer |
| Chemiluminescence western blotting | Antibodies for signal enhancement |
| Fluorescent western blotting | |
Technical Resources | About us | Contact us | Bulk Service
Licenses | Conditions of Use | Privacy Policy | Ordering Information

FM 545248