

"I have used a wide variety of secondaries and Jackson ImmunoResearch has consistently been the best. The fluorophores are bright and stable and their selective (x reactivity removed) secondaries have always shown species specificity in multiple labeling."
Janet Duerr, Ohio UniversityRating: 5.0
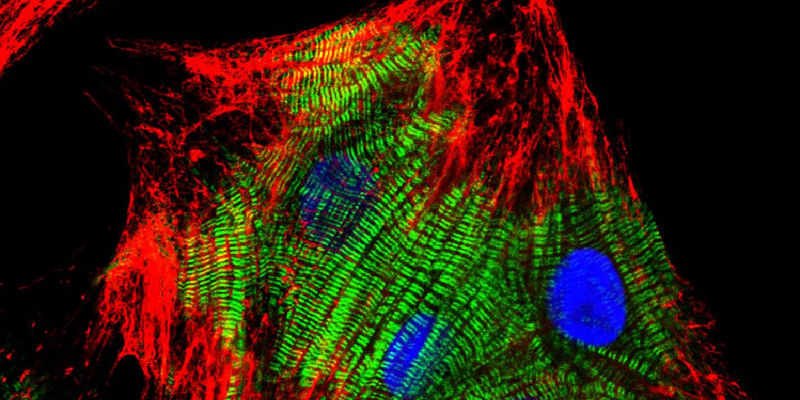
Lymphatic endothelial cells in culture. Cell-cell adhesion proteins stained red (using VEcadherin) and F-actin in cyan (phalloidin). Nuclei are yellow (counterstained with DAPI). Jackson ImmunoResearch products: Cy™3 AffiniPure Donkey Anti-Goat IgG (H+L) (min X) (705-165-147).
Credit: Derek Sung
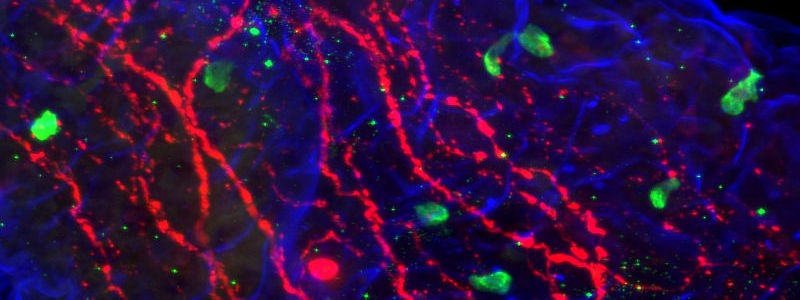
Immunohistochemistry (IHC) and Immunofluorescence immunostaining are powerful techniques, indispensable in research and in clinical diagnostics. They are a staple of the pathology lab for disease diagnostics and classification. In research, Immunostaining techniques are used to interrogate many biological processes, including visualization of protein expression patterns, characterization of protein interactions, and identification of tissue boundaries. The researcher can thereby observe the distribution and localization of specific structures within the context of the cellular architecture.
A tissue analyte is specifically recognized by a primary antibody, which may be directly conjugated (direct immunostaining); or the primary may itself be detected by a conjugated secondary antibody (indirect immunostaining), allowing visualization of the analyte. The conjugated antibody can be labeled with any of several reporter molecules, including enzymes, fluorophores and colloidal gold. The expression of multiple analytes can be observed and characterized by individual primary/secondary antibody pairs in multiple labeling protocols.
Analytes of interest may be detected on the surface of the tissue or interrogated internally through permeabilization of the sample. The analyte (antigen) can be detected directly (Figure 2 A), or indirectly (Figure 2 B) using a fluorescent (immunofluorescence) or reporter enzyme (colorimetric) probe conjugated to a secondary antibody.
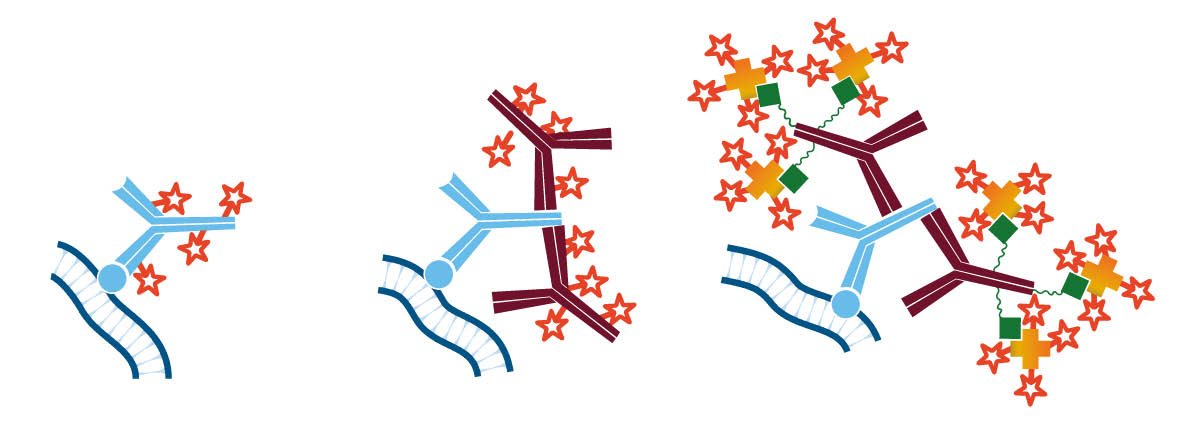
The indirect method using a secondary antibody offers many advantages. It preserves the antigen binding site on the primary antibody by conjugating to the secondary antibody. Since secondary antibodies are available conjugated to a wide variety of fluorophores, the indirect method enhances the possibilities for multiple labeling. In addition, it provides inherent signal enhancement, whereby multiple secondary antibodies bind to one antigen-bound primary antibody. Signal can be further enhanced by using a biotinylated secondary antibody followed by conjugated streptavidin (Figure 2 C), or by using the peroxidase-anti-peroxidase (PAP) method.
Factors that contribute to the success of an experiment are the tissue of interest, primary antibody choice, secondary antibody specificity, and options for signal detection. Proper blocking assures optimal signal-to-noise ratio, and control proteins aid in the interpretation of results.
The source (species) of experimental sample will influence the choice of antibodies. Certain tissue types contain endogenous immunoglobulin (Ig), and others exhibit vasculature in which remnant blood may provide endogenous Ig. For indirect immunostaining, the secondary antibody must recognize the specific primary antibody but not endogenous Ig. It is most convenient to use a primary antibody that is derived from a different host animal than the tissue of interest: in this case select a secondary antibody that has been cross-adsorbed (min X) to minimize recognition of endogenous Ig. If the experimental strategy includes using a primary antibody from the same host species as the tissue of interest, endogenous Ig can be masked by blocking with monovalent Fab fragments of secondary antibodies. It is also possible to label primary antibodies with Fab fragments prior to incubation with the sample.
Read more about Fab fragment secondary antibodies.
Tissue fixation is the process of stabilizing and preserving a specimen in a configuration that approximates its natural state. As the innate aqueous environment is altered by chemical fixation, an antigenic site may be modified so that it cannot be recognized by a primary antibody that would detect its native state. Many primary antibodies are marketed as “validated” to perform under specified fixation conditions, though this information may not be available. In some cases, antigen retrieval protocols can return antigenic sites to their native state.
Note that the concept of antibodies validated to perform in immunostaining is unique to primary antibodies. If a primary antibody binds its target site, a labeled secondary antibody will bind to it regardless of tissue fixation.
Tissue samples may reveal several types of intrinsic signal. Autofluorescence (signal in the absence of fluorescent probe molecules) may be evident in some regions of the spectrum. When planning an immunofluorescent protocol, observe an unlabeled tissue sample using available filter sets. Choose fluorophores that are compatible with spectral regions that have limited autofluorescence. It is possible to suppress autofluorescence with various reagents, though the efficiency of these treatments varies depending on both tissue type and fixation method.
Intrinsic signal may also derive from endogenous enzymes and/or biotin. To test for and control these types of background signal, see our Blocking and Controls guide.
The primary antibody should recognize the protein of interest under the intended experimental conditions. Some primary antibodies have been validated for use under specific conditions, e.g. fixation methods. An antibody that has been validated only for Western blotting may not be appropriate for immunostaining.
Polyclonal antibodies recognize multiple epitopes, maximizing the opportunity for the antibody to detect the antigen, but may result in detection of homologous proteins and lead to background signal. Monoclonal antibodies consist of identical immunoglobulin molecules produced from a single cell line, and are appropriate when the detection needs are epitope specific. However, they may give a weaker signal than polyclonals. Controls can be used to help determine the suitability of antibodies for each assay system.
Ideally, the host species of the primary antibody should be a different species from the sample species to avoid cross-reactivity with endogenous proteins. If the primary antibody and the tissue of interest are the same species, blocking protocols can be used to prevent off-target signal.
Multiple labeling is used for the detection of more than one analyte in the same assay. Primary antibodies derived from different species, or mouse monoclonal antibodies of different subclasses, are optimal choices for multiple labeling. If these choices are unavailable, multiple labeling can be achieved through special strategies.
Primary antibodies may detect the target protein satisfactorily for one experimental protocol, but not work as well under different conditions. It is good practice to optimize and therefore validate the antibody each time the experimental conditions are altered, for example, when the tissue type is changed or a different secondary antibody detection system is used.
When selecting secondary antibodies for IHC and multiple labeling experiments, consider the criteria in the following table.
The affinity-purified antibodies marked "ML" (multiple labeling) have been specifically prepared to meet these criteria.
Caution: Avoid the possible formation of immune complexes by diluting antibodies in PBS/Tween only (without carrier proteins such as normal serum). To limit unwanted interactions incubate reagents sequentially rather than mixing multiple antibodies.
| Requirement | Antibody | Example |
|---|---|---|
| The secondary antibody must not recognize endogenous tissue immunoglobulins. | Choose a secondary antibody cross-adsorbed against the species of interest. | In Rat tissue, choose Goat Anti-Mouse IgG (H+L)(min X Hu, Bov, Hrs, Rb, Rat Sr Prot) |
| For visualizing a primary antibody on tissue from the same species (e.g. mouse on mouse labeling) block endogenous Ig with Fab fragments. | See Fab fragment blocking protocols. | |
| The secondary antibody must not bind to endogenous Fc receptors and proteins. | Block with normal serum from the same species as the secondary antibody. | See normal serums. |
| Multiple secondary antibodies must not recognize one another | Use secondary antibodies that are derived from the same host species if possible. | Donkey Anti-Mouse Donkey Anti-Rabbit Donkey Anti-Guinea Pig |
| Each secondary antibody must only recognize its target primary antibody. | For multiple labeling protocols, choose secondary antibodies which have been cross-adsorbed against the other species employed in the experiment. | Antibodies raised in donkeys have been highly cross-adsorbed against other species. |
| For labeling different mouse subclasses, use subclass specific antibodies. | Goat Anti-Mouse IgG1, IgG2a, etc. See anti-Mouse IgG subclass specific antibodies. | |
| If appropriate cross-adsorbed antibodies are unavailable use a Fab protocol for multiple labeling. | See Fab fragment blocking protocols. |
Analytes can be visualized under a fluorescence microscope by using a fluorescent conjugate, or under a light microscope by using an enzyme conjugate followed by a chromogenic substrate (colorimetric detection). Detection by light microscopy is also possible using silver enhancement of a colloidal gold antibody.
Each detection method can offer advantages, in terms of sensitivity, quantification, cost-per-assay or capacity for detection of multiple analytes. Table 1 below compares the two detection methods.
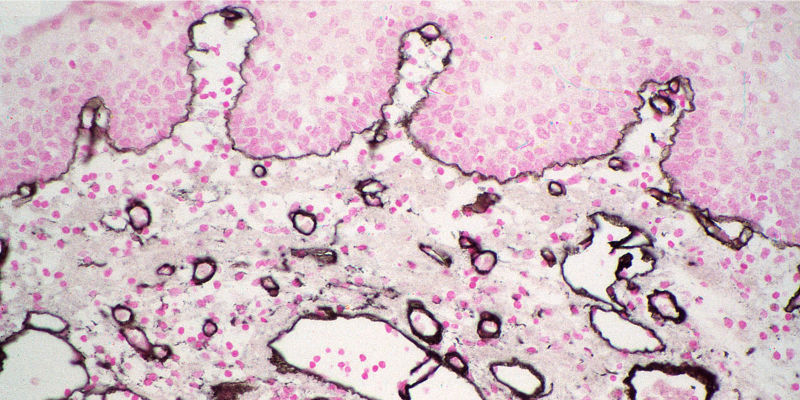
Alkaline Phosphatase (AP) and Horseradish Peroxidase (HRP) conjugates can be used for colorimetric detection. Colorimetric detection can offer quick and easily obtained results, with excellent detection sensitivity. Multiple analyte detection can be achieved by using different chromogens in sequential incubations.
Read more about reporter enzymes.
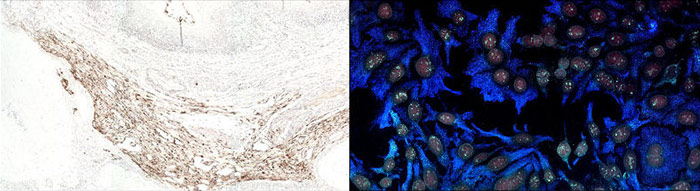
Immunofluorescence uses a fluorescent dye conjugate to visualize antigens. Fluorescent IHC allows for detection of multiple analytes, and up to 5 color immunofluorescence is possible using the appropriate fluorescent dyes and filter sets. Stripping and reincubating with additional antibodies has resulted in 20 analytes being probed on a single tissue sample.
Read more about fluorescent probes.
| Colorimetric | Fluorescent | |
|---|---|---|
| Sensitivity | HRP conjugates are more sensitive than fluorescent conjugates. | Fluorophores such as Alexa Fluors® and Cyanine dyes are brighter and more photostable than FITC, increasing their sensitivity. |
| ImmunoGold with silver enhancement is also very sensitive. | ||
| Expense and ease | Simple light microscopes are commonly available. | Epifluorescent microscopes are readily available and versatile. Expensive multichannel microscopes may require use of core facilities, increasing cost and difficulty of experiment. |
| Resolution | Detail and depth of interrogation are limited by the quality and thickness of the samples. | Maximum detail can be detected using confocal microscopy, including compilation of 3D images using scanning laser techniques to visualize within much thicker sample slices. |
| Electron microscopy using ImmunoGold reagents offers excellent resolution. | Super-resolution microscopy allows images to be resolved down to 50 nm. | |
| Multiplexing (simultaneous detection of multiple antigens) | Multiple analytes can be labeled by using different chromogens for detection in sequential incubations. | Up to 5 color immunofluorescence can be performed with appropriate microscope laser and filter set up. Additional multiplexing has been reported using rounds of stripping and reincubating. |
| Colocalization | Analytes can be identified by channel, and their signals merged to identify overlap by color. | |
| Color stability | Stains are stable and do not fade during examination under the microscope. Slides can be stored for years. | Photostability can be variable across fluorescent conjugates, making data collection time sensitive. Permanent mounting media can increase longevity of archived slides. |
| Advanced techniques | Fluorescent dyes can be used for a number of techniques to characterize biological interactions. FRET (Förster Resonance Energy Transfer) can be used to examine structural characteristics, binding stoichiometries, and affinities. Super-resolution microscopy allows images to be resolved down to 50 nm. Fluorescent reagents make it possible to observe live cells in real time. |
|
Blocking steps can dramatically improve results and simplify analysis of immunostaining experiments. To avoid unexpected signal from non-specific, conserved sequence and/or Fc-receptor binding, we recommend blocking with normal serum from the host species of the labeled antibody (5% v/v). Other blocking reagents, such as BSA or commercially available blockers, may also be effective at minimizing background.
The addition of experimental controls will improve analysis of results and aid troubleshooting.
Read more about blocking and controls.
Suggested dilution ranges for Immunostaining techniques such as Immunohistochemistry (IHC), Immunocytochemistry (ICC), and Immunofluorescence (IF) are shown in the table below.
| Product | Conjugate | Histo-/Cyto chemistry |
|---|---|---|
| Whole IgG and F(ab')2 secondary antibodies | Unconjugated | 10-20 µg/ml |
| Fab secondary antibodies | Unconjugated | 20-40 µg/ml |
| Whole IgG, F(ab')2 and Fab fragment secondary antibodies | Alexa Fluor® 488, 594, 647, DyLight™ 405, and Cy™3 | 1:100-1:800 |
| Whole IgG, F(ab')2 and Fab fragment secondary antibodies | AMCA, BV421™, BV480™, Cy2, FITC, TRITC, RRX, and Texas Red | 1:50-1:200 |
| Whole IgG secondary antibodies | Cy™5 | 1:100-1:400 |
| Whole IgG and F(ab')2 secondary antibodies | Horseradish Peroxidase | 1:500-1:5,000 |
| Whole IgG, F(ab')2 and Fab secondary antibodies | Biotin-SP (using Fluorophore-Conjugated Streptavidin) | 1:200-1:1,000 |
| Whole IgG and F(ab')2 secondary antibodies | Biotin-SP (using enzyme-Conjugated Streptavidin) | 1:500-1:5,000 |
| Streptavidin | Horseradish Peroxidase | 1-2 µg/ml |
| Streptavidin | Alkaline Phosphatase | 1-2 µg/ml |
| Streptavidin | Alexa Fluors®, DyLight 405, and Cy3 | 0.5-2 µg/ml |
| Streptavidin | Cy5 | 0.5-2 µg/ml |
| Streptavidin | All Other Fluorophores | 2-5 µg/ml |
| 4 nm Gold-Antibody Complexes | 1:20-1:200 | |
| 6, 12 nm Gold-Antibody Complexes | 1:20-1:40 | |
| 18 nm Gold-Antibody Complexes | 1:10-1:20 | |
| Peroxidase-Anti-Peroxidase (PAP) | 25-50 µg/ml | |
| FabuLight™ Anti-IgM | All Fluorophores | To complex with primary antibody in solution, use 1:1 weight ratio of Fab: primary antibody (15:1 molar ratio). |
| FabuLight Anti-IgG | All Fluorophores | To complex with primary antibody in solution, use 1:1 weight ratio of Fab: primary antibody (3:1 molar ratio). |
| Normal Serum | 5% (v/v) for blocking | |
| ChromPure™ | 10 µg/ml |
